


Here comes the under 5 vaccine! But I have questions...

Si quiere leer la versión en español, pulse aquí.
Today, Pfizer announced they plan to submit an Emergency Use Authorization (EUA) application for the COVID19 vaccine for children under 5 years of age. Here is my best attempt to explain what is (and is not) known about the bumpy regulatory road to authorization for our youngest kiddos.
What’s happened leading up to now?
The entire process for a vaccine under 5s started back in May 2021. Since then, Pfizer has gone through several necessary steps to get the vaccine approved. I talked about this extensively in a previous post, but here’s a recap:
May 2021: Pfizer’s clinical trials for the 6 months to under-5 population began.
This was almost a year after the adult clinical trials began, which is a typical “age de-escalation” design: We test adults before we test more vulnerable populations like children.
Phase I/IIa: Pfizer started with the first step: Phase I “dose finding” study where they tested several different dosages: 3 µg, 10 µg, 20 µg, and 30 µg (adult dosage) among a few dozen healthy children. Their ultimate goal was to find the “sweet spot”—the lowest dosage possible causing the lowest rate of side effects, but also enough dosage to generate an immune response. In other words, what’s the minimum dosage with just enough benefit? Pharmaceutical companies have different approaches to finding the sweet spot for vaccines. For example, while Moderna has been trying to push the red line (what’s the maximum dosage without having too much risk?), Pfizer has had the opposite approach (what’s the minimum dosage with just enough benefit?). As you can imagine, each approach comes with advantages and disadvantages.
Nonetheless, Pfizer announced during an investor call (see slide below) that the 3 µg (lowest dosage) worked. They also decided not to move forward with 10 µg because of higher rate of fevers among 2-<5 years.
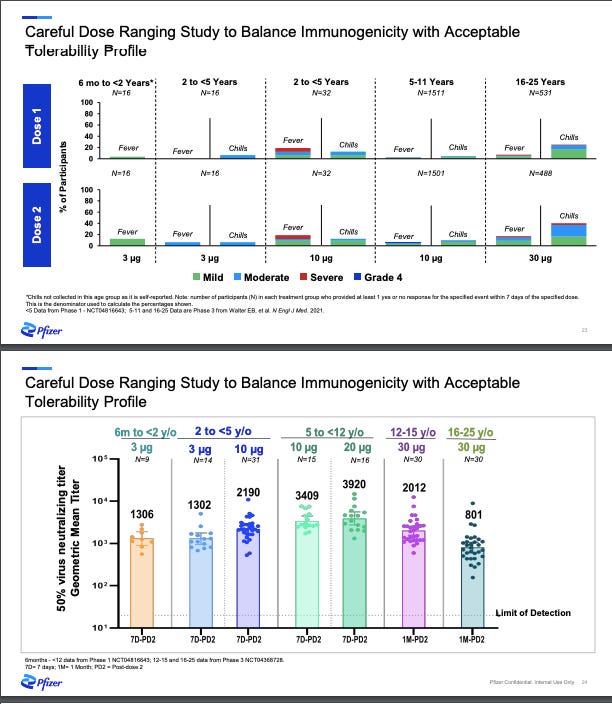
Phase IIb/III: The next phase of the clinical trials is where a much larger group—a couple thousand kids—is given this “sweet spot” dosage (3 µg). By increasing the number of children, scientists confirm the dosage, but also have more “power” to detect rare side effects and improve the generalizability of the vaccine impact by including a wide range of ages, races/ethnicities, children with comorbidities, etc. At this stage Pfizer also evaluated vaccine effectiveness by randomizing children to the vaccine or a placebo. This allowed scientists to evaluate a pre-determined “primary outcome” called immunobridging. This was not efficacy because we already know the biotechnology worked from our adults trials. Pfizer just needed to make sure that a smaller dose worked just as well: Does the smaller dosage (3 µg) mount the same immune response among under 5 year olds compared to the full dosage (30 µg) among 16-25 year olds?
December 2021: Pfizer announced results of their Phase II/III trial. There were two big wins and one massive set back:
Wins: Vaccine was safe, so no severe events occurred. The vaccine dosage worked for kids between 6 months and under 2 years.
Failure: Vaccine dosage didn’t work for 2- to <5-year-old population
Because of the failure, the process for an EUA stopped. It was even stopped for those under 2 years (although the vaccine dosage did work) because of the nature of de-escalation design: The FDA doesn’t jump age groups. Pfizer announced their Plan B: The same kids from the Phase II/III trial get a third dose (3 µg) 2 months after the second dose. They expected results by mid-2022.
What happened today?
Pfizer announced that they will seek an EUA for the first two doses (3 µg) for <5 year olds. Pfizer doesn’t have data from the third dose among this cohort yet because this takes time (they’re continuing trials to get this data eventually). If there is no new data since the last press release (December 2021), then what changed? I don’t know. But, if we read in-between the lines, we can assume it centers around Omicron:
The Phase IIb/III trial used same outcome as for the 5-11 year old trial: neutralizing antibodies. Neutralizing antibodies are our first line of defense and help prevent infection altogether—they act really fast so the virus doesn’t enter our cells and thus it doesn’t replicate, so we don’t get symptoms, don’t become contagious, and don’t spread the virus.
Before Omicron, neutralizing antibodies were working really well. With Omicron, though, our first line of defense is largely eroded without a booster. Thankfully, our secondary line of defense (T-cells) still works. This is largely why partially-vaccinated people continue to stay out of the hospital. So this begs the question: Are neutralizing antibodies still a fair measure to indicate that a vaccine is useful? Or, should we evaluate T-cell activation or, more broadly, efficacy against hospitalization and death? Discussions have likely been happening behind closed doors at the FDA.
All while Omicron infiltrated unvaccinated pockets, including children, like we’ve never seen before. From today’s press release, it was clear this was a big driving factor to push for an EUA, as it is “in response to the urgent public health need in this population”. In addition, the virus continues to mutate. Together, this changing landscape is pushing the FDA to reconsider the goalpost mid-game.
What happens next?
Pfizer’s application for EUA needs to be officially submitted to the FDA in the coming days. The FDA will then review the data, which takes about 4-6 weeks.
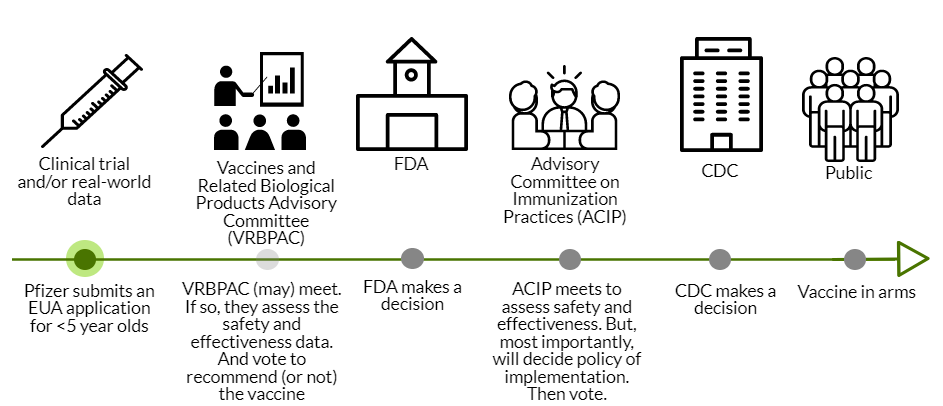
Next, the FDA could call VRBPAC (Vaccines and Related Biological Products Advisory Committee). VRBPAC is an external scientific committee that would review the data to give their opinion about the safety and effectiveness of the vaccine for under 5 year olds. This step was not conducted for the adolescent Pfizer vaccine; VRBPAC doesn’t have to be called for an EUA amendment. If VRBPAC is called, this would make for a very interesting discussion, as this committee made it clear in previous meetings that they want clear, rigorous data to make decisions. Only two voting members have verbally recognized that we sometimes have to make decisions based on limited data. VRBPAC could decide (through a majority vote) that we don’t have enough justification to authorize <5’s the vaccine. The FDA makes the final decision, but rarely (if ever?) do they go against VRBPAC’s vote. There is a lot of power at play here.
CDC Review: If the FDA doesn’t call on VRBPAC or VRBPAC votes that this vaccine is safe and effective, then this goes to the ACIP (an external scientific committee for the CDC). ACIP assesses safety and effectiveness, too. But their key role is policy: who gets the vaccine and how, global equity considerations, etc. For example, ACIP could decide that the vaccine should only be available to the <2 year olds. Or they could comment on off-label usage. ACIP is amazing at conducting rigorous risk-benefit analyses, even with limited data. So, we’ll get a fantastic dive into the benefits and risks of their proposed policy. ACIP will then vote on whether to recommend the vaccine for dissemination in the United States. Then the CDC Director makes the final recommendation.
In all, if all goes well (which is not guaranteed) we could have vaccines in arms by mid-March.
Many questions
As you can tell, there is very limited data and little transparency thus far. So, going forward, we all need the following four key questions answered:
What was the original Phase III data for <5s? How far off were the neutralizing antibodies for 2-<5 year olds? If this was very far off, how comfortable do we feel with just vaccinating and hoping that the third dose will work?
Is there a new goalpost, like T-cells or hospitalizations? And, why was it chosen? If it’s hospitalizations, does Pfizer have enough sample size to measure effectiveness confidently given that hospitalizations are more rare for kids?
What is the safety profile of the proposed dose for kids under 5?
What is the plan if a third dose does not work? Is Pfizer concurrently testing other doses? And, what are the implications for long COVID and MIS-C, in particular, if the third dose doesn’t work?
Bottom line: I hope I didn’t make this more confusing, but… it’s complicated. As a mom with two kids under 5, I couldn’t be more excited that a vaccine continues to move forward. As an epidemiologist and public health official, I have lots of questions. We will be getting answers in the weeks to come as our checks and balances play out. I’ll be reporting back, like always, with the cliff notes. Hang in there!
Love, YLE
EDIT: It looks like VRBPAC has already scheduled a meeting for Feb 15. So, this is the next step in the process! We will know a whole lot more then.
“Your Local Epidemiologist (YLE)” is written by Dr. Katelyn Jetelina, MPH PhD—an epidemiologist, biostatistician, professor, researcher, wife, and mom of two little girls. During the day she has a research lab and teaches graduate-level courses, but at night she writes this newsletter. Her main goal is to “translate” the ever-evolving public health science so that people will be well equipped to make evidence-based decisions. This newsletter is free thanks to the generous support of fellow YLE community members. To support the effort, please subscribe here:
Looks like VRBAP has already been scheduled for 2/15. Does this change your thinking about the likely timeline and/or likelihood of approval?
I just want to say thank you. As a mom of an under 2 and as an elementary school educator, I am so grateful for your ability to break down the process and explain the variables. As soon as I heard this news today I was anxiously awaiting your post to walk through the details. Thank you.